Der Name der Firma Pfizer ist verbunden mit Skandalen und Machenschaften, die man fast als Markenzeichen dieser Firma ansprechen könnte.
Das glauben die meisten Menschen erst einmal gar nicht und halten das für „Verschwörung“ oder auf neuhochdeutsch: „fake news“.
Aber in den letzten Jahren hatte ich dazu bereits einiges berichtet und veröffentlicht – alles mit Quellenangaben:
Daher ist es kaum verwunderlich, dass auch in Sachen „Coronaimpfungen“ und deren angebliche Wirksamkeit die Firma einiges zu bieten hat, nur nichts Gutes für sein Klientel, die „Geimpften“:
- Corona-Impfstoffe: Vergessen Sie die „Jubelberichte“
- Die Wirksamkeit der RNA- und DNA-basierten „Impfungen“
Es ist kein Zufall, dass Impfstudien seit Menschengedenken praktisch ohne Placebokontrolle durchgeführt werden:
Eine solche Praxis erlaubt mehr Raum für großzügige Interpretationen in Bezug auf Wirksamkeit und Sicherheit:
Die Daten von Pfizer für deren „Coronaimpfung“ standen von Anfang an im Kreuzfeuer der Kritik, wie zum Beispiel von BMJ-Co-Herausgeber Peter Doshi[1], der nicht nur die trickreich errechnete Wirksamkeit von 95 % kritisierte, sondern auch auf fehlende Daten und etliche Widersprüche hinwies.
Von daher gab es schon früh etliche Vermutungen, dass es in der Pfizer-Zulassungsstudie deutlich mehr Todesfälle in der Gruppe der „Geimpften“ gab als angegeben wurde. Grund hierfür: Frisierte Daten um eine Zulassung zu erreichen. Auch das ist nicht neu, wie ich bereits hier berichtete: Verfälschte Studien für schnellere Zulassung
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Neue Studie – neue Erkenntnis
Am 4. September 2023 erschien eine noch nicht peer-reviewte Arbeit[2] [3], die angibt, dass viele Todesfälle in der Pfizer-Zulassungsstudie erst nach dem Datenstichtag (data cutoff) gemeldet wurden.
Selbiger Stichtag wurde für die Erstellung einer Informationsbroschüre verwendet, die wiederum Einzug in das „Zulassungsverfahren“ hielt, was darin mündete, dass die Mortalitätsdaten bei der Entscheidung für oder gegen eine Zulassung nicht berücksichtigt wurden.
Oder mit anderen Worten: Aufgrund dieser Daten hätte der „Impfstoff“ nie zugelassen werden dürfen.
Insgesamt handelt es sich um 38 Todesfälle – 21 Todesfälle in der Verumgruppe („Geimpfte“) und 17 Todesfälle in der Placebogruppe („Ungeimpfte“, die nur physiologische Kochsalzlösung erhalten hatten). Wie es aussieht, wusste Pfizer über diese Todesfälle Bescheid. Denn die Autoren vermerken Folgendes:
„In vielen dieser 38 Fälle unterstützte die im CRF (clinical report files) bereitgestellte Dokumentation die Todesursachendiagnose nicht ausreichend oder erlaubte es nicht, die Möglichkeit eines kardiovaskulären Ereignisses durch eine Autopsie auszuschließen.
Die häufige Kommunikation zwischen den Ärzten von Pfizer/BioNTech und dem medizinischen Personal der Prüfzentren ist aus den CRFs ersichtlich, die oft recht umfangreich waren, einige weit über 400 bis 900 Seiten.“
Die Autoren sehen ihre Arbeit insofern als einmalig an, da sie sich als die erste Gruppe bezeichnen, die eine Analyse der Originaldaten durchgeführt haben, ohne mit dem Sponsor der Studie, Pfizer und BioNTech, verbunden zu sein.
Sie betrachten ihre Arbeit als eine forensische Analyse von besagten 38 Teilnehmern, die zwischen dem 27. Juli 2020, was den Beginn der Phase 3 der klinischen Studie markiert, und dem 13. März 2021, dem Enddatum des 6-Monats-Zwischenberichts, verstorben sind.
Hinweis zwischendurch: Im April 2020 wollten die EMA und Pfizer die Phase 3 der Studie einfach ganz weglassen. Das gelang zwar nicht, aber schon vor dem Beginn der klinischen Tests kam vom RKI eine unerwartete Schützenhilfe: Von dort warfen Wissenschaftler in die Debatte, eine Impfpflicht für Menschen in medizinischen und pflegerischen Berufen anzuordnen.
Im Abstract heißt es dann weiter, dass es Unstimmigkeiten zwischen den Ergebnissen im Pfizer-Bericht und dem von den Pfizer-Studienleitern verfassten Zwischenbericht gegeben hatte. Hier gab es nämlich einen 3,7-fachen Anstieg der Todesfälle aufgrund von kardiovaskulären Ereignissen in der Verumgruppe im Vergleich zur Placebogruppe. Und genau das wurde von Pfizer tunlichst verschwiegen.
Folgende Grafik aus der Studie zeigt, dass schon ab Herbst 2020 mehr Probanden in der Verumgruppe starben als in der Placebogruppe:
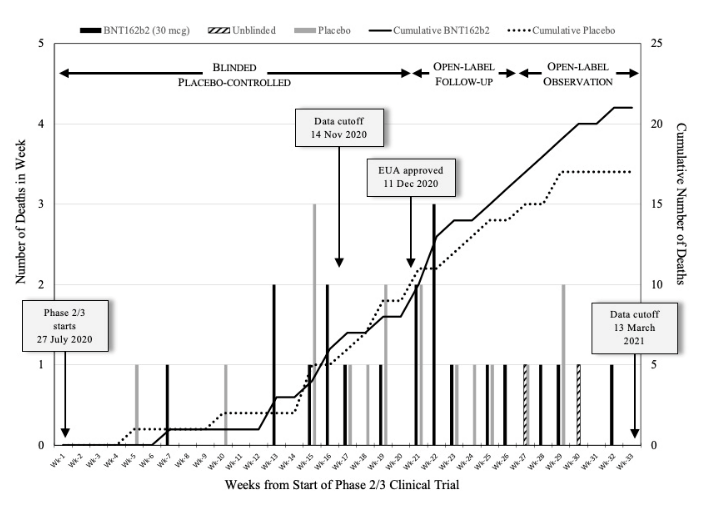
Die schwarzen Balken repräsentieren die Fälle der Verumgruppe, die grauen Balken die der Placebogruppe. Die schwarze Linie zeigt die kumulative Inzidenz für die Todesfälle in der Verumgruppe, die gestrichelte Linie die der Placebogruppe.
Bereits hier zeigte sich deutlich ab, dass es die erwartete geringere Sterblichkeit unter der „Impfung“ nicht nur nicht gab, sondern dass hier eine Tendenz zu beobachten war, die auf eine höhere Sterblichkeit unter der neuen „Impfung“ deutete.
Noch mehr Vertuschung?
Folgende Tabelle zeigt die veröffentlichten „tollen“ Daten von Pfizer (linke Seite) und die Daten vom 6-Monats-Zwischenreport (rechte Seite):

Unschwer lässt sich hier erkennen, dass die Zahl der Todesfälle für den Zeitraum vom Beginn der Studie bis zum 14. November 2020[4] fast halbiert wurde, mit vier Todesfällen für die Placebogruppe und nur zwei Todesfälle für die Verumgruppe beim Pfizer-Report.
Der Zwischenreport weist jedoch für den gleichen Zeitraum elf statt sechs Todesfälle aus – fünf Todesfälle in der Placebogruppe und sechs Todesfälle in der Verumgruppe. Also hat Pfizer bei der Veröffentlichung der Daten vier Todesfälle in der Verumgruppe unterschlagen. Das Gleiche gilt auch für kardiovaskuläre Probleme. Hier berichtete der Pfizer-Report nur einen Fall in der Verumgruppe, während der Zwischenreport von vier Fällen redet.
Da stellt sich die Frage, ob diese Beobachtungen der ausschlaggebende Grund waren, die Studie bereits nach wenigen Monaten zu entblinden und die Teilnehmer in der Placebogruppe ebenfalls zu „impfen“, da man es ethisch nicht vertreten konnte, ihnen den „95-prozentigen Schutz“ vorzuenthalten?
Denn die Zahlen im Zwischenreport geben Grund für die Vermutung, dass bei einer Fortführung der Studie mit einer Placebogruppe die Schere zwischen Verum-Todesfällen und Placebo-Todesfällen noch weiter aufging und damit die Zulassung des Produkts gefährdete.
Oder oder mit anderen, brutalen Worten: Pfizer brauchte in der Placebogruppe mehr Todesfälle, um wenigstens nicht den Verdacht aufkommen zu lassen, dass seine Gen-Injektionen für die Todesfälle verantwortlich gemacht werden konnten. Und diese Todesfälle konnte man nur produzieren, indem man auch die Teilnehmer in der Placebogruppe mit den Gen-Injektionen versah.
Saubere Arbeit! Toll!
Das Endresultat für den Zeitraum von Juli 2020 bis Mitte März 2021 sieht so aus:
Laut Pfizer gibt es 34 Tote – 18 in der Verumgruppe und 16 bei Placebo.
Der Zwischenbericht spricht von 38 Todesfällen – 21 in der Verumgruppe und 17 bei Placebo.
Fazit
In den ersten siebeneinhalb Monaten der Zulassungsstudie zeigte sich nicht nur, dass es für die Gen-Injektionen von Pfizer/BioNTech keine 95-prozentige Wirksamkeit gibt, sondern dass es auch den Schutz vor Tod nicht nur nicht gibt, sondern dass diese Spritzen die Wahrscheinlichkeit für ein Versterben erhöhen.
Darum darf man getrost davon ausgehen, dass die Voraussetzungen für eine Zulassung nicht auch nur eine Sekunde gegeben waren. Und es dürfte als erwiesen gelten, dass die Daten offensichtlich bewusst gefälscht wurden, um diese Zulassung nicht zu gefährden. Dieses ist ein weiteres „Meisterstück“ von Pfizer in Sachen „tarnen und täuschen“. Die Zulassungsbehörden haben sich entweder dumm gestellt oder waren mit von der Partie. Wie dem auch sei, es ist Zeit für Konsequenzen aus diesen Erkenntnissen.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter dazu an:
Dieser Beitrag wurde am 14.10.2023 erstellt und letztmalig am 24.08.2024 aktualisiert.
Quellen:
- [1] Peter Doshi: Pfizer and Moderna’s “95% effective” vaccines—we need more details and the raw data – The BMJ
- [2] Forensic Analysis of the 38 Subject Deaths in the 6-Month Interim Report of the Pfizer/BioNTech BNT162b2 mRNA Vaccine Clinical Trial[v1] | Preprints.org
- [3] Michels-preprints202309.0131.v1-2.pdf
- [4] Der „Datenstichtag“, nach dessen Ergebnisse die „Notfallzulassung“ zusammengeschustert wurde. Die Daten nach dem Stichtag spielten für die Entscheidung keine Rolle mehr, obwohl sich hier massive Probleme abzeichneten.