„Impfen lassen oder auf Beatmung verzichten“. Diese Aussage war die Schlagzeile der BILD vom 19.12.
Die Aussage stammt von Prof. Dr. Henn. Herr Henn ist Mitglied im Ethikrat der Bundesregierung. Diese „Meinung“ von Herrn Henn verbreitete sich ziemlich rasch. Diese Sache zeigt sehr gut, auf welchem „Niveau“ wir angekommen sind.
https://freie-impfentscheidung.de/wp-content/uploads/2020/12/ethikrat-henn-impfen-min.png346610rgraeberhttps://freie-impfentscheidung.de/wp-content/uploads/2025/07/impflogo.jpgrgraeber2020-12-27 10:09:452022-08-18 19:39:19Ethikrat Mitglied Prof. Henn: „Impfen lassen oder auf Beatmung verzichten“
Die FDA hat den neuen RNA-„Impfstoff“ von Pfizer/BioNTech am 11. Dezember 2020 als Notzulassung registriert.
Die FDA ist die Food and Drug Administration (FDA) der USA. Auf deutsch: US-Behörde für Lebens- und Arzneimittel, zuständig für die Lebensmittelüberwachung und Arzneimittelüberwachung und Zulassung. Als solche ist sie dem amerikanischen Gesundheitsministerium unterstellt.
Die FDA schreibt in einem Merkblatt zur Zulassung des Pfizer-BioNTech COVID-19-Impfstoffs:
Der Pfizer-BioNTech COVID-19-Impfstoff wurde nicht derselben Art von Überprüfung unterzogen, wie ein von der FDA zugelassenes oder freigegebenes Produkt. Die FDA kann eine EUA erlassen, wenn bestimmte Kriterien erfüllt sind, welche umfassen, dass keine angemessenen, zugelassenen, verfügbaren Alternativen vorhanden sind. Zusätzlich dazu basiert die Entscheidung der FDA auf der Gesamtheit der verfügbaren wissenschaftlichen Nachweise, dass das Produkt während der COVID-19-Pandemie zur Vorbeugung von COVID-19 wirksam ist und dass die bekannten und potentiellen Vorteile des Produkts die bekannten und potentiellen Risiken des Produkts überwiegen. Alle diese Kriterien müssen erfüllt werden, damit das Produkt zur Behandlung von Patienten während der COVID-19-Pandemie eingesetzt werden darf.
Die EUA für den Pfizer-BioNTech COVID-19-Impfstoff ist in Kraft für die Dauer der COVID-19 EUA-Erklärung, die eine Notfallverwendung dieser Produkte rechtfertigt, außer diese wird beendet oder zurückgezogen (wonach die Produkte nicht länger verwendet werden dürfen). Quelle: https://www.fda.gov/media/144617/download
Die neuartige Impfung wurde nicht nur in einem Schnellverfahren entwickelt, sondern es gibt auch eine Notzulassung. Dr. Peter Doshi hat dazu noch weitere Analysen der Zulassung erstellt. Aber wer ist eigentlich Dr. Doshi?
Wer ist Dr. Doshi?
Dr. Doshi ist ein kritischer Beobachter der Impfindustrie, und das nicht erst seit die neuen RNA-„Impfungen“ auf dem Programm stehen. Ich hatte im Dezember 2013 einen Beitrag über die Grippeimpfung geschrieben. Und auch hier zitierte ich seine bereits kritische Stellungnahme zu der Effektivität und Verträglichkeit von Grippeimpfungen: Grippeimpfung – nichts als ein schlechter Marketingwitz?
Bereits damals sah er es als erwiesen an, dass die Studien, auf denen die Zulassungen von Grippeimpfungen beruhen, kaum wissenschaftlichen Standards genügten.
Dr. Doshi ist einer der Editoren der Fachzeitschrift BMJ (British Medical Journal). Er ist Assistenzprofessor für pharmazeutische Gesundheitsforschung an der Universität von Maryland. Hier forscht er nach Richtlinien in Bezug zur Medikamentensicherheit und deren Umsetzung im Zusammenhang mit Zulassungsverfahren, evidenzbasierter Medizin und Diskussionen über Daten und deren Verfügbarkeit.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Die seltsamen Phänomene einer Notfallzulassung
Peter Doshi und Matthew Herder kamen zu dem Schluss, dass die amerikanische „Food and Drug Administration“ die wichtigste Entscheidung in ihrer Geschichte in diesem Jahr gemacht hat – die Notfallzulassung für die Pfizer/BioNTech Covid-19-Impfung.
Was dabei auffiel ist, dass die Zulassungsbehörde offensichtlich nur einen einzigen Prüfer jeweils für die beiden wissenschaftlichen Schlüsseldisziplinen abgestellt hatte (Klinik und Statistik). Und es fiel auf, dass die Durchsicht und Bewertung der Unterlagen in nur drei Wochen erfolgte, ein Vorgang, der sonst eine Reihe von Monaten in Anspruch nimmt.
Nach der Notfallzulassung durch die FDA zogen dann auch Großbritannien und Kanada nach. Die FDA hat bislang den Ruf, der „Goldstandard“ in Sachen Zulassung von Medikamenten und Medizinprodukten zu sein. Dieser Ruf rührt auch daher, dass die FDA bislang (in der Regel) rigorose Zulassungsanforderungen aufrecht erhielt.
Doshi und Herder fahren weiter fort, dass die FDA sehr wahrscheinlich die einzige Zulassungsbehörde in der Welt ist, die dauerhaft Patientendaten von klinischen Studien sammelt und diese mit den Daten für Zulassungen abgleicht. Für derartig gründliche Analysen benötigt die FDA normalerweise rund zehn Monate. Hier sind dann eine Reihe von Experten mit den verschiedenen Analysen beschäftigt, als da sind: Klinische Medizin, Statistik, Pharmakologie, Chemie, Pharmakovigilanz (Medikamentensicherheit/Nebenwirkungsprofil) und andere mehr.
Zusammengenommen ergeben diese Analysen ein „Aktionspaket“, welches laut Gesetz nicht später als 30 Tagen nach der Zulassung öffentlich gemacht werden muss.
Da wir es allerdings jetzt mit einer „Pandemie“ zu tun haben, erfolgte der Review der Pfizer-Impfung viel schneller als gewöhnlich. Hauptgegenstand der Analyse war die werkseigene Phase-3-Studie mit 44.000 Teilnehmern. Die FDA Analytiker absolvierten ihre Arbeit in nur drei Wochen (20. November bis 11. Dezember).
Bei dieser monumentalen Aufgabe erhebt sich sofort die Frage, warum die FDA mit nur so wenigen Experten die Arbeit hat durchführen lassen? Warum hat sie nicht mehr Experten zum Einsatz kommen lassen?
Laut Zulassungsprotokoll der FDA (siehe Link oben) gibt es einige wissenschaftliche Disziplinen, wie zum Beispiel die Pharmakovigilanz, die von mehreren Analytikern bearbeitet wurden. Aber die beiden Disziplinen, die mit der Bewertung der klinischen Studiendaten und deren Ergebnisse beauftragt waren, die Analytiker für Klinik und Statistik, hatten wohl ihre Aufgaben alleine zu bewältigen.
Und dies ist auf mindestens zwei Gebieten vollkommen unzureichend. Erstens ist es unverständlich, wie eine Arbeit von mehreren Monaten ohne zusätzliche Analytiker auf nur 22 Tage (Samstage und Sonntage eingeschlossen) komprimiert werden konnte. Denn eine gründliche Analyse beschäftigt sich mit der Bewertung der patientenbasierten Datenlage, was auch die Nachverfolgung und Analyse von individuellen Fallberichten und die unabhängige Analyse von Rohdaten involviert.
Vor der „Pandemie“ war es nicht ungewöhnlich, nur einen einzigen Experten-Namen für die jeweilige Disziplin zu benennen. Aber bei dem, was momentan auf dem Spiel steht und in Anbetracht der kurzen Entwicklungszeit der neuen Impfung, denken Doshi und Herder, dass die FDA einen deutlich gründlicheren Job abliefern würde als üblich. Und genau das scheint nicht der Fall zu sein.
Fragen zur Pfizer-Studie
Aus wissenschaftlicher Sicht gibt es mehrere große Fragezeichen, die diese „Studie“ auszeichnen. Es beginnt damit, dass in der Verumgruppe (die Gruppe, die die echte Impfung bekommen hatte) drei bis viermal mehr Fieber- und Schmerzmittel eingenommen hatte als die Teilnehmer der Placebogruppe.
Dies alleine ist schon eine aus wissenschaftlicher Sicht unzulässige Veränderung des Studiendesigns, um die Zahl der Nebenwirkungen in der Verumgruppe zu reduzieren.
Gleichzeitig erfolgt hiermit eine Aufhebung der Verblindung der Studie, denn man kann mit der Gabe der Medikamente deutlich bestimmen, welcher Teilnehmer zu welcher Gruppe gehört (die mit Schmerzmitteln zur Verumgruppe; die ohne zur Placebogruppe). Damit kann diese Arbeit auf keinen Fall den Anspruch einer „doppelblinden“ Studie erheben.
Und genau das wäre doch bei einer neuen Form der „Impfung“ von besonderer Wichtigkeit, um deren wissenschaftliche Absicherung zu gewährleisten.
Die FDA und ihre Experten haben jedoch diesen beiden Umständen so gut wie keine Beachtung geschenkt. Zumindest gibt es in deren Report keine Anhaltspunkte dafür.
Die FDA reagiert
Es kam eine umgehende Antwort von Dr. Peter Marks, dem Direktor des „Zentrums zur Evaluierung und Erforschung von Biologika“ der FDA. Diese Abteilung der FDA ist für die Sicherstellung der Sicherheit, Reinheit, Wirksamkeit etc. von Biologika und verwandten Produkten, inklusive Impfungen, verantwortlich. Dr. Marks ist hämatologischer Onkologe.
Der bezeichnete die „Meinungsäußerung“ (wie oben beschrieben) von Doshi und Herder als inkorrekt und die Arbeit der Experten in der FDA im Zusammenhang mit der Notfallzulassung der Pfizer-Impfung als falsch charakterisiert. Angeblich würde diese „Meinungsäußerung“ der beiden Autoren nicht zeigen, dass sie die Analysearbeit des FDA-Teams verstehen würden.
Die Agenturangestellten hätten über Monate rund um die Uhr gearbeitet, sogar nachts, an Wochenenden und Feiertagen. Und das wäre erfolgt, lange bevor der Antrag auf die Notfallzulassung eingereicht wurde. Und auch der Vorwurf, es wären zu wenig Experten bei der Analyse der eingereichten Daten involviert gewesen, wäre haltlos, da 100 Agenturangestellte, inklusive aus dem leitenden Management, an der Arbeit beteiligt gewesen wären.
Die Autoren würden deutlich zeigen, dass sie nicht verstehen, dass es sich hier nicht um eine wissenschaftliche Publikation handele. Die aufgeführten Namen seien lediglich den entsprechenden verantwortlichen Leuten je nach Disziplin zuzuordnen. Sie würden nicht das gesamte Team von aufopferungsbereiten Mitarbeitern repräsentieren, die unermüdlich gearbeitet hätten, um tiefgehende Analysen für die Notfallzulassung zu erarbeiten.
Er fährt weiter fort, dass es sich hier um eine heroische Tat handele, bei der in nur 22 Tagen die Notfallzulassung erarbeitet werden konnte. Und dabei hätte die FDA alles erdenklich Mögliche getan und jeden Stein umgedreht.
Und weil die Pandemie so viel Leid verursacht hätte, wären die Analyseexperten mit einem erhöhten Verantwortungsbewusstsein und besonderer Sorgfalt bei ihrer Arbeit vorgegangen. Dabei wären die strengen wissenschaftlichen Maßstäbe, die Amerika und die Welt von der FDA erwarten, in jeder Hinsicht erfüllt worden. Jede anderslautende Vermutung sei eine Beleidigung der unglaublichen Bemühungen.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Die Antwort auf die Antwort
Doshi und Herder bemerken, dass Marks ihnen vorwirft, dass sie das ernst nehmen, was im Zulassungsprotokoll geschrieben steht. Und da steht, wie weiter oben schon ausgeführt, nur ein Name für die klinische und nur ein Name für die statistische Evaluierung.
Weiter würde Marks behaupten, dass die FDA-Mitarbeiter schon seit mehreren Monaten arbeiteten. Daran gäbe es keinen Zweifel. Nur die Phase 3 Studie ist erst vor wenigen Wochen beendet und deren Resultate am 20. November der FDA überreicht worden. Das wiederum heißt, dass trotz monatelanger Arbeit die Analyse der Studiendaten nur auf drei Wochen beschränkt war. Und das sei Lichtgeschwindigkeit im Vergleich mit dem normalerweise monatelangen Evaluierungsprozess der FDA.
Weiter gibt Marks keine Beispiele an, wie die Basisdaten während dieser 22 Tage kritisch analysiert wurden. Von daher glauben Doshi und Herder nach wie vor, dass eine gründliche Analyse und Auswertung dieser Daten in dieser kurzen Zeit unmöglich sei, und schon gar nicht mit nur einem Experten für die klinische und einem Experten für die statistische Analyse.
Auch die Angabe von Marks, dass mehr als 100 Angestellte hier involviert gewesen wären, könnte man glauben. Aber Doshi und Herder wundern sich, warum bei der Pharmakovigilanz dann zwei Namen gelistet sind, und bei der Chemie, Herstellung und Kontrolle sind es sogar drei Namen, aber nur jeweils ein Name für die Klinik, Biostatistik und Toxikologie.
Ich frage mich auch, ob diese „formalen“ Unregelmäßigkeiten bei der Darstellung der Ergebnisse nicht auch Ausdruck für tiefergreifende Unregelmäßigkeiten beim Zulassungsprozedere sein könnten?
Doshi und Herder fahren fort, dass die Frage nach der Menge der Experten die eine Seite der Medaille sei. Die andere Seite ist die Qualität der Analysen und der sich daraus ergebenden Notfallzulassung. Sie führen das bereits weiter oben kurz andiskutierte Argument über die mögliche Entblindung der Studie aufgrund der gegebenen Nebenwirkungen und der Gabe der Fieber- und Schmerzmedikamente an. Hier gibt es keine Stellungnahmen seitens der FDA in ihrem Report.
Doshi und Herder fragen sich auch, wie viele Leute mit einer unabhängigen wissenschaftlichen Beurteilung der Daten beauftragt worden waren. Sie glauben auch nicht, dass es möglich gewesen sei, dass abweichende Meinungen, die die Qualität der Zulassungsstudie infrage stellten, frei dokumentiert und diskutiert werden konnten.
Fazit
Eine Zulassungsstudie für eine neuartige „Impfung“ im Eilverfahren, ohne wirkliche doppelblinde Kontrolle, mit massivem Einsatz von Schmerzmitteln in der Gruppe der Geimpften, die trotz dieser Maßnahme signifikant mehr Nebenwirkungen (im doppelstelligen Prozentbereich) aufzeigt als die Placebogruppe (was offensichtlich nicht zur Kenntnis genommen wird), wo aber Infektionsraten von nur 0,08 % in der Impfgruppe und 0,74 % in der Placebogruppe und deren Differenz als 95-prozentige „Wirksamkeit“ als signifikant erachtet wird, ist das Dokument dafür, dass jede Form von Wissenschaftlichkeit über Bord geworfen wurde.
Diese Form der „Wissenschaft“ hatte sich bereits seit Jahren entwickelt und abgezeichnet, wobei man zum damaligen Zeitpunkt noch bemüht war, durch die Vermeidung von Angaben von Interessenskonflikten eine gewisse wissenschaftliche Fassade aufrechtzuerhalten. Doch diese Fassade ist heute nicht mehr nötig, wie diese Notfallzulassung in beeindruckender Art und Weise dokumentiert.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Wer über Impfung und Impfschutz redet, der geht von der uralten Hypothese aus, dass eine Impfung das Immunsystem des Geimpften zu einer Antikörper-Produktion anregt, die sich gegen das Antigen oder Teile des Erregers richten. Und wenn dann der natürliche Erreger auftritt, dann wäre der Geimpfte aufgrund seiner bereits bestehenden Antikörper vor selbigem geschützt.
Wenn diese Hypothese stimmen würde, dann wäre dies eine tolle Angelegenheit. Was sich jedoch in den letzten Jahren immer mehr abzeichnet ist, dass diese Hypothese vielleicht für einige Fälle/Erreger richtig zu sein scheint. Aber für andere Erreger ist sie vollkommen falsch. Warum?
Beispiel: Dengue-Fieber.
Das Dengue-Virus ist wie SARS-CoV-2 ein RNA-Virus. Auch hier gab es bereits eine Reihe von Bemühungen, Impfungen zu entwickeln, die jedoch mit einer Reihe von Problemen aufwarteten (um es vorsichtig auszudrücken):
Man hatte hier bei der Impfung beobachtet, dass die Geimpften nach einer Impfung viel schwerere Krankheitsverläufe bei einer zweiten Infektion durchmachen mussten als Personen, die keine Impfung erhalten hatten. Es hatte sich auch gezeigt, dass diese Impfung keinen Schutz vor einer Infektion darstellt. Der einzige, für mich fragwürdige, Bonus ist, dass Personen, die bereits eine Infektion durchgemacht haben und die dann eine Impfung erhalten, möglicherweise einen milderen Verlauf durchmachen.
Das dreht die Welt der Impf-Hypothese vollkommen auf den Kopf. Derzufolge soll doch eine Impfung vor einer Infektion schützen. Und jetzt muss man erst einmal eine Infektion durchgemacht haben, damit man vor der Impfung geschützt ist.
Welcher Teufel im Detail hält hier die schulmedizinische Wissenschaft und die Doktrin von der heilbringenden Impfung zum Narren?
Impfung ADE
Die Abkürzung „ADE“ bedeutet nicht „Tschüss“, obwohl die dahinter verborgenen Mechanismen momentan kaum eine andere Möglichkeit bereithalten, als sich an den Gedanken zu gewöhnen, sich von der Impfung zu verabschieden.
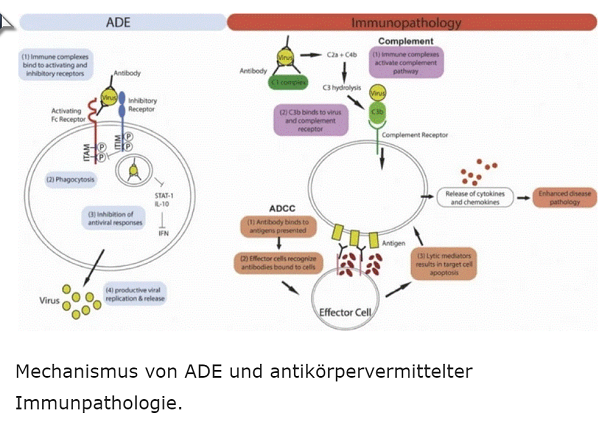
ADE steht für „antibody dependent enhancement“, also „infektionsverstärkende Antikörper“.
Es scheint also zwei verschiedene Arten von Antikörpern zu geben, die von der klassischen Impf-Hypothese überhaupt nicht berücksichtigt werden.
Hier werden nur die „neutralisierenden Antikörper“ berücksichtigt, die im Falle einer Infektion in der Lage sind, die entsprechenden Pathogene zu binden und zu neutralisieren. Über diesen Mechanismus kann eine Vermehrung der Pathogene unterbunden werden, was darin resultiert, dass der Betroffene nicht erkrankt (= keine Symptome zeigt).
Aber diese Form der Abwehr funktioniert auch nur dann, wenn genügend neutralisierende Antikörper produziert werden können, die der aufgenommenen Viruslast mengenmäßig entsprechen. Ist die Viruslast größer als die produzierte Menge an Antikörpern, dann kann es dennoch zur Erkrankung kommen.
Die nicht-neutralisierenden Antikörper, die ADE, bewirken das komplette Gegenteil. Vereinfacht beschrieben binden die Erreger an diese Antikörper, worauf dieser Komplex erst in der Lage ist, über bestimmte Rezeptoren auf der Zelloberfläche in die fragliche Zelle vor- und einzudringen, die elementare Voraussetzung für Viren, um sich reproduzieren zu können. Und je mehr ADE gebildet werden, desto effektiver verläuft dieser Prozess, was in noch schwereren Krankheitsverläufen mündet.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
ADE und noch einmal ADE
Diese nicht-neutralisierenden Antikörper haben noch ein weiteres Ass im Ärmel: Sie verursachen eine Immunpathologie. Über eine Reihe von Mechanismen führen sie zu einer übermäßigen Aktivierung des Immunsystems, die von einer überschießenden Freisetzung von Zytokinen und Chemokinen gekennzeichnet ist.
Dieser Zytokinsturm führt in den betroffenen Geweben zu massiven Entzündungsreaktionen mit dementsprechend hohen Konzentrationen an freien Radikalen, die das Gewebe so nachhaltig schädigen können, dass es untergeht und zu Langzeitschäden oder sogar lebensbedrohlichen Situationen führt.
Diese Mechanismen können bei einer natürlichen Infektion auftreten, und, das ist die wahrscheinlichere Variante, auch oder gerade bei einer Impfung, die darauf ausgelegt ist, vermeintlich schützende Antikörper zu produzieren.
Bei der Dengue-Impfung ist, wie weiter oben beschrieben, diese paradoxe Reaktion bereits beschrieben.
Covid-19-Impfung ADE?
Der Mechanismus, den ich gerade für die Dengue-Impfung beschrieben habe, scheint auch für Impfungen gegen Coronaviren zu gelten. Denn es ist kein Zufall, dass bis auf den heutigen Tag noch keine Coronaviren-Impfung entwickelt werden konnte. Grund hierfür sind ADE, die in einer Reihe von Tierversuchen mit und an verschiedenen Tierspezies beobachtet wurden und in vielen Fällen für die geimpften Tiere mit dem Tode endeten.
Hierbei handelt es sich allerdings nicht um eine neue Erkenntnis, sondern ist aufgrund der jahrzehntelangen Bemühungen, Impfungen gegen Coronaviren zu entwickeln, der fundamentale „Stolperstein“ für die Entwicklung einer solchen Impfung.
Eine Arbeit aus dem Jahr 2003 kennzeichnete bereits die Bedeutung und Funktion von ADE und die Tatsache, dass der Mechanismus, der diese unvorteilhafte Form von Antikörpern entstehen lässt, nicht nur nicht verstanden wird, sondern die Entwicklung von zuverlässigen und sicheren Impfungen nahezu unmöglich zu machen scheint.
Es gibt ein Interview mit Robert Kennedy Jr. vom Mai 2020, wo dieser die Geschichte der Entwicklung von Coronavirus-Impfstoffen zusammenfasst und erläutert. Die in diesem Zusammenhang durchgeführten Experimente schienen wohl in den USA so gefährlich gewesen zu sein, dass im Jahr 2014 der damalige Präsident Obama einen Stopp der Entwicklung anordnete.
Federführend bei der Entwicklung in den USA war bis zu diesem Zeitpunkt Dr. Fauci, der danach nicht mit der Entwicklung aufhörte, sondern einfach seine Entwicklungsbemühungen in das berühmt-berüchtigte Labor von Wuhan verlegte.
So erfahren wir in diesem Interview, dass um das Jahr 2012 rund 30 Coronavirus-Impfungen entwickelt worden waren. Von diesen 30 Impfungen wählte man die vier aussichtsreichsten Kandidaten und produzierte die entsprechenden Impfungen, die dann an Frettchen ausgetestet wurden. Frettchen sind in diesem Zusammenhang eine beliebte Tierspezies, da sie im Bereich von Lungenerkrankungen den pathophysiologischen Vorgängen beim Menschen am nächsten kommen.
Ergebnis dieser Bemühungen: Alle vier Impfungen waren in einem hohen Maße in der Lage, Antikörper gegen Coronaviren zu bilden. Das alleine reicht bereits aus, um eine Impfung von den Behörden zugelassen zu bekommen. Also alles in bester Ordnung?
Danach kam die „kalte Dusche“. Denn als die Frettchen mit natürlichen Coronaviren konfrontiert wurden, entwickelten alle einen schweren Krankheitsverlauf und verstarben. Grund hierfür waren massive Entzündungsreaktionen in allen Organen, die zu einem multiplen Organversagen führten. Die Ursache für dies alles waren ADE.
Weiter erfahren wir hier die traurige Realität von Menschenversuchen, wieder einmal. Denn bereits in den 1960er Jahren hatten Wissenschaftler versucht, einen Impfstoff gegen RSV (Respiratorisches Synzytial Virus – mit gleicher Symptomatik wie Influenza und Corona) zu entwickeln. Dabei hatte man die Austestung an Tieren einfach übersprungen und den Impfstoff sofort an Kindern ausprobiert.
Auch hier geschah das Gleiche wie bereits beschrieben. Bei rund 35 Kindern entwickelte sich eine hervorragende Antikörperlage. Als die Kinder von natürlichen RS-Viren infiziert wurden, wurden alle schwerst krank und zwei verstarben sogar in der Folge. Daraufhin wurde die Entwicklung dieses Impfstoffs verworfen.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Der gegenwärtige Stand der Impfstoffentwicklung
Zu dieser Frage hatte ich bereits einiges an Beiträgen herausgebracht:
Ein sehr kritischer Beitrag von Peter Doshi im „British Medical Journal“ charakterisiert den Wahnsinn, der bei der Impfstoffentwicklung gegen SARS-CoV-2 die Oberhand gewonnen zu haben scheint. Wie bereits von vielen vorausgesagt, gibt es inzwischen zu wenig Covid-19-Fälle, an denen eine Impfung zuverlässig ausgetestet werden könnte.
Laut Herrn Doshi sehen die Studienprotokolle von Pfizer, Moderna und AstraZeneca so aus, dass in Ermangelung von schweren Verlaufsformen bereits milde Symptome und/oder ein positiver PCR-„Test“ ausreichen, um Aspiranten für eine Teilnahme an der Studie zu qualifizieren.
Die kritischen Fälle bei einer Covid-19 Infektion sind derartig gering, dass man auf diese Verlegenheitslösung ausweichen muss, um überhaupt eine Studie machen zu können. Die amerikanische CDC hatte im April veröffentlicht, dass nur 3,4 % aller Infizierten mit Symptomen einen Krankenhausaufenthalt benötigten. Hier gab es eine altersbedingte Variationsbreite: 1,7 % in der Altersgruppe zwischen null und 49 Jahren; 4,5 % bei einem Alter zwischen 50 und 64 Jahren und 7,4 % in den Jahren 65 und darüber.
Und da die meisten Leute mit einer symptomatischen Covid-19-Infektion nur sehr milde Symptome entwickeln, gibt es für Studien mit selbst über 30.000 Teilnehmern keine nennenswerten Zahlen mit einer für die Studie gewünschten schweren Symptomatik.
Wofür ist dann eine solche Studie gut? Zur was taugt sie?
Aus wissenschaftlicher Sicht sind diese Studien vollkommen unsinnig. Dies gibt sogar der leitende Mediziner von Moderna zu, indem er einräumt, dass die Studie, die gerade von der Firma durchgeführt wird, keine statistische Power (Aussagekraft) in Bezug auf die zu untersuchenden Ergebnisse hat.
Auch hierüber hatte ich bereits berichtet (hier noch einmal der Link dazu). In der Pfizer-Studie gab es nur 94 Fälle bei 43.000 Teilnehmern. Wo soll da eine statistische Relevanz herkommen? Bei der Moderna-Studie sieht es nicht viel besser aus. Dafür verhindern diese neuen Impfungen erst nach 255 Impfungen eine Infektion (rein statistisch gesehen!). Die anderen Geimpften haben von dieser Impfung keinen Nutzen, dafür aber das höchstwahrscheinliche „Privileg“ von schwerwiegenden Nebenwirkungen.
Wie diese aussehen und wie hoch sie ausfallen, das können Sie in dem zuletzt genannten Beitrag (siehe Link) nachlesen.
Und somit hat man diese Studien so ausgelegt, dass sie einfach Erfolge zeigen müssen. Wie macht man so etwas? Man erklärt einfach, dass das primäre Ziel die relative Risiko-Reduktion von mindestens 30 % der Teilnehmer ist, die eine Covid-19-Infektion entwickeln.
Und das hat man bei Pfizer zum Beispiel bereits realisiert, denn hier zeigte sich eine relative Reduktion von knapp 91 %. Hurra!
Denn es hatte sich gezeigt, dass die Infektionsrate in der Impfgruppe bei 0,04 % lag, die in der Placebogruppe bei 0,43 %. Damit ergibt sich eine absolute Risiko-Reduktion von 0,39 %. Kann man das als eine exorbitant hohe Reduktion bezeichnen? Vielleicht in der „neue Normalität“? In der Alten jedenfalls würde man eine solche Zahl als nicht aussagekräftig und damit als signifikanzlos werten.
Berechnet man allerdings den prozentualen Anteil von 0,39 % aus 0,43 %, dann erhält man seine gewünscht hohe Zahl von knapp 91 % relativer Risiko-Reduktion. Toll!
Angesichts dieser desaströsen Zahlen scheint man dazu übergegangen zu sein, um überhaupt noch ein Produkt verkaufen zu können, neue Argumente für die Impfung aus dem Boden zu stampfen. Und das ist ausschließlich die Behauptung, dass eine Impfung schwere Krankheitsverläufe verhindern könnte. Wie bitte?
Die Impfung ist nicht in der Lage, Infektionen zu verhindern, die Mortalitätsrate unter Infektionen zu senken und die Geimpften zu schützen? Wozu dann die Impfung? Wer entwickelt schwere Krankheitsverläufe? Sind dies nicht nur im Wesentlichen Menschen mit Vorerkrankungen im Bereich chronischer Leiden?
Wieso benötige ich eine solche Impfung, wenn ich als gesunder Mensch eine minimale Wahrscheinlichkeit habe, überhaupt eine Infektion zu bekommen, und wenn, dann nur mit milden oder gar keinen Symptomen?
Karl Lauterbach hat ja auch vor laufender Kamera verkündet, dass diese Impfung nicht das Ende von den „AHA“-Regeln bedeutet, obwohl Spahn und Merkel vor nicht allzu langer Zeit behauptet hatten, dass nur eine Impfung dies alles beenden kann. Aber ich denke, dass wir auch hierbei davon ausgehen dürfen, dass es sich wieder mal um eine dicke Lüge gehandelt hat.
Impfung ja – „AHA“-Regeln jetzt erst recht! Was hat das mit der Gesundheit der Bevölkerung zu tun? Es könnte deutlich mehr mit der „Kontrolle“ der Bevölkerung zu tun haben (wie es ja manche seit Monaten postulieren), sowie den Profiten der Pharmaindustrie, die durch die Impfung auf sie warten.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Gegen den Widerstand gegen die Impfung
Diese Maßnahmen gegen den Widerstand machen sich inzwischen in allen Bereichen des Lebens bemerkbar. Und man wird es kaum glauben: Sie sind auch bei den Studien selbst auszumachen.
Ende Oktober 2020 erschien im „International Journal of Clinical Practice“ ein Beitrag, der aufzeigte, dass die momentan durchgeführten Covid-19-Studien am Menschen einen weiteren Baustein unethischer Praxis implementiert haben. Bei Studien müssen die Studienteilnehmer detailliert auf mögliche Risiken und Nebenwirkungen der Substanzen hingewiesen werden, die an ihnen ausprobiert werden.
In diesen Studien wird den Teilnehmern jedoch verschwiegen, dass die Gefahr von ADE (siehe Diskussion weiter oben) besteht, die mit schweren Erkrankungsverläufen oder sogar dem Tod enden könnten. Dadurch wird eine informierte Zustimmung seitens der Teilnehmer unmöglich gemacht.
Warum wird dies gemacht? Die Erklärung ist denkbar einfach: Bei einer voll umfassenden Erklärung der Situation dürfte es einen signifikanten Prozentsatz von Teilnehmern geben, die dann dankend auf eine Teilnahme verzichten. Das würde die Durchführung von solchen Studien noch mehr gefährden als dies ohnehin schon der Fall ist. Also lässt man alle Ethik sausen und macht, was das Produktmanagement der Pharmafirmen von ihnen verlangt.
Fazit
Die neuen Corona-Impfungen versprechen eine höchst unangenehme Veranstaltung zu werden. Auf der einen Seite eine sehr fragliche Effektivität, die auf dem Papier durch statistische Tricks herbeigerechnet wird, auf der anderen Seite die Wahrscheinlichkeit von möglichen massiven und lebensbedrohlichen Nebenwirkungen.
Wenn man dann noch mit in Betracht zieht, dass mit den RNA-Impfungen eine vollkommen neue Form der „Impfung“ eingesetzt wird, die noch weniger erprobt ist als die klassischen Formen von Impfungen, dann dürfte klar sein, dass eine so schnelle Zulassung solcher „Impfungen“ die „Pandemie“ nur als Vorwand nimmt, um in einem noch nie da gewesenen Ausmaße Kasse zu machen.
https://freie-impfentscheidung.de/wp-content/uploads/2020/08/spritze-impfung-123rf-90739460-Olga-Yastremska-min.jpg565848rgraeberhttps://freie-impfentscheidung.de/wp-content/uploads/2025/07/impflogo.jpgrgraeber2020-12-23 13:42:042022-08-18 19:39:19Covid-19-Impfung und Immunsystem
Die Impfung gegen Covid-19 hat begonnen. In den USA, Großbritannien, Kanada, Bahrain, Saudi-Arabien und Mexiko ist der Impfstoff von Pfizer/BioNTech offiziell zugelassen worden und auch in Deutschland wurde mit dem Impfen begonnen. Und kaum ist die Impfung auf dem Markt, kommen auch schon die ersten Meldungen über schwere Nebenwirkungen.
Ich kann mich nicht erinnern, dass bei anderen neu auf den Markt gekommenen Impfungen seinerzeit so schnell und so häufig Nebenwirkungen gemeldet wurden.
Wie sieht es momentan aus?
Mitte November 2020 erschien in dem „British Medical Journal“ ein Beitrag, der aufgrund der spärlich veröffentlichten Daten zu diesem Zeitpunkt die Pfizer-Impfung einzuschätzen versuchte. Er bestätigte für die Impfung zwar eine Relative Risiko-Reduktion von 90 %, die aber auf einer Absoluten Risiko-Reduktion von nur 0,4 % beruhte.
Das heißt, dass so wenig Teilnehmer in der Studie an Covid-19 erkrankten, dass man unter normalen Umständen von einer Impfung absehen müsste. Dementsprechend hoch fiel auch die „Number Needed to Vaccine“ (NNV) (Impfungen nötig, um eine Infektion zu verhindern) aus, nämlich 256 Impfungen, um eine Infektion zu verhindern.
Pfizer war die 90-prozentige Relative Risiko-Reduktion wohl nicht hoch genug, sodass die Firma einige Daten veröffentlichte, aus denen dann eine 95-prozentige Reduktion abzulesen war. Die Absolute Risiko-Reduktion stieg auf 0,71 % an, was immer noch kein Grund ist, über Impfungen nachzudenken. Und die NNV sank von 256 auf 141. Auch diese neue Zahl spricht nicht gerade von einem überragenden Nutzen der Impfung.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Auf der anderen Seite häufen sich die Berichte von Nebenwirkungen der RNA-„Impfung“, und das bereits nach rekordverdächtig kurzer Zeit. Inzwischen kursiert ein Video von einer Krankenschwester im Netz, die kurz nach der Impfung während einer Pressekonferenz ohnmächtig wird. Ein kommentierter Beitrag von „RT.de“ berichtet hier von Dementis, dass diese Ohnmacht etwas mit der Impfung zu tun haben könnte.
Sie selbst solle gesagt haben, dass sie leicht in Ohnmacht falle, wenn sie Schmerzen verspüre. Wenn das stimmt, warum ist sie dann nicht schon während der Impfung in Ohnmacht gefallen, wo sie den Stich erhalten hatte?
Im gleichen Artikel wird berichtet, dass kurz nach der Zulassung in Großbritannien die britischen Aufsichtsbehörden einen Warnhinweis herausgaben. Demzufolge sollten Personen mit einer „signifikanten Vorgeschichte allergischer Reaktionen“ die Impfung vermeiden. Und sie berichten weiter, dass mindestens zwei Beschäftigte im Gesundheitswesen von Alaska schon nach der ersten Impfdosis (die Pfizer-Impfung sieht zwei Impfungen vor) „schwere Nebenwirkungen“ bekommen hätten.
Dieser Bericht wurde von der „Epoch Times“ bestätigt. Hier erfahren wir, dass es sich um eine Anaphylaxie handelt, die für die eine Betroffene eine intensivmedizinische Behandlung notwendig machte. Die Frau ist mittleren Alters und hatte zuvor keine Allergien gehabt. Der andere Betroffene hingegen konnte nach rund einer Stunde die Notaufnahme schon wieder verlassen.
Der Artikel spricht auch noch von zwei weiteren Fällen, zwei Frauen, aus Großbritannien, die nach der Impfung starke allergische Reaktionen gezeigt hatten. Die beiden Frauen hatten allerdings eine Vorgeschichte für Allergien gegen Eiweiß und Medikamente. Dies scheint die Ursache für den Warnhinweis aus Großbritannien zu sein, bei bestehender Vorgeschichte für Allergien die Impfung zu vermeiden.
Die englischsprachige Webseite „Life Site“ konkretisiert seine Argumente, denen zufolge ein unverhältnismäßig hoher Anteil an Geimpften hohes Fieber (38 Grad und höher) entwickeln, nämlich 15,8 % in der Altersgruppe zwischen 18-55 Jahren. Fast die Hälfte der Geimpften (45 %) mussten Medikamente gegen Fieber oder Schmerzen nehmen. Über die Hälfte (55 %) zeigten Kopfschmerzen und 62 % Fatigue. Seine Schlussfolgerung: „Nein, wirklich, das ist zu viel. Vielleicht gibt’s da ein Problem …“
Mehr Literatur zum nachdenklich werden
Die vorausgesagten Probleme mit dieser neuartigen RNA-„Impfung“ scheinen sich viel schneller zu bewahrheiten als befürchtet. Ich hatte hierzu eine Reihe von Beiträgen verfasst:
Die „Wissenschaftliche Gesellschaft für Quantenmedizin und Bewusstseinsforschung“ hat einen interessanten Blog auf ihrer Webseite herausgebracht, der sich mit der Coronaimpfung kurz aber umfassend beschäftigt. Es werden hier die zentralen Fragen gestellt, die jeden bei der Entscheidungsfindung helfen sollen, sich für oder gegen die Impfung zu entscheiden. Die Fragen beziehen sich nicht nur auf die Coronaimpfung, sondern sollten auch bei/vor jeder anderen Impfung gestellt werden.
Teil 4 der Erörterung bezieht sich auf die Situation, wo jemand sich für die Impfung entschlossen hat. Auch hier sollten aus Sicherheitsgründen eine Reihe von Fragen beachtet und beantwortet werden.
Weil eine Impfung einen massiven Eingriff in die Physiologie darstellt, der mit entsprechenden Folgen verbunden sein kann, sollte ein Arzt…
– immer eine ausführliche Anamnese (Vorgeschichte) durchführen
– die Körperfunktionen untersuchen
– Laborbefunde erheben, als da sind Blutbild, Blutchemie, Immunstatus, Hormonstatus, Schilddrüsenfunktion etc.
– auf mögliche Allergien testen, insbesondere gegenüber Inhaltsstoffen des Impfstoffs
– die Medikamente, die der Patient zum Zeitpunkt der Impfung nimmt, erfassen und berücksichtigen
– diese Medikamente auf Wechselwirkungen mit den Inhaltsstoffen und Zusatzstoffen der zu verabreichenden Impfung überprüfen.
Es bleibt in diesem Zusammenhang auch zu fragen, ob die für Deutschland vorgesehenen Massen-Impf-Stationen diesen Fragen gerecht werden? Ich schätze, dass es spätestens bei den Laborbefunden als Antwort ein klares „NEIN“ geben wird. Und auch die Erfassung der Medikamente, die Allergietestung und die Überprüfung auf Wechselwirkungen dürfte kaum gewährleistet sein.
Und auch die sonst verpflichtende Erklärung von Nebenwirkungen und Risiken der Impfung durch den behandelnden Arzt dürfte unter diesen Bedingungen, wenn überhaupt, dann nur extrem kurz und wenig informativ (schon gar nicht abschreckend) ausfallen.
Es kommt noch dazu, dass für die RNA-„Impfung“ keine Aufklärung über Spätfolgen gemacht werden kann, da es keine Untersuchungen zu Spätfolgen gibt bzw. geben kann. Denn diese Impfungen sind in Windeseile aus dem Boden gestampft worden. Die Spätfolgen werden wir in dem jetzt eingeleiteten Großversuch am Menschen sehen und zu spüren bekommen.
Und zum schlechten Schluss stellt sich auch die Frage nach der Notfallbehandlung, die, wie weiter oben bereits diskutiert, bei den Beispielen aus Alaska und Großbritannien kurz nach der Impfung notwendig wurde? Gibt es in diesen Massen-Impf-Stationen gut ausgestattete Intensiveinheiten, die Notfälle vor Ort versorgen können?
Damit bleibt für den Impfwilligen die ernsthafte Frage, ob er/sie sich wirklich diesem Risiko aussetzen möchte, zumal die Notwendigkeit einer Impfung sogar jetzt auch von den Fachleuten, die keine Impfgegner sind, ernsthaft infrage gestellt wird.
Von daher betrachtet auch die „Gesellschaft für Quantenmedizin“ die anberaumten Massenimpfungen als „menschenrechtsverletzende Verantwortungslosigkeit“, da keine der notwendigen ethischen Voraussetzungen und verpflichtenden Sicherheitsmaßnahmen erfüllt werden. Das ist die schulmedizinische Form der „Massenmenschhaltung“.
Trotz fehlender Haftung
Die Industrie hat ihren „Freifahrtschein“ in Sachen Haftung. Wenn Sie sich dennoch entschließen sollten, sich dieser Impfung zu unterziehen, dann sollten Sie sich zumindest Folgendes notieren:
– Name, Adresse, Zulassung des Arztes, der Sie geimpft
– ist der Arzt selbstständig oder ist er in einer Praxis angestellt ist beziehungsweise bei wem ist er angestellt?
– wer ist Ihr möglicher Ansprechpartner, falls nach der Impfung Probleme auftreten? Wo und wann ist der Ansprechpartner erreichbar?
– wer haftet bei Impfschäden, da die Industrie keine Haftung übernimmt?
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
https://freie-impfentscheidung.de/wp-content/uploads/2024/05/KI_Virus_Corona_Impfung_Spritze.jpeg10241024rgraeberhttps://freie-impfentscheidung.de/wp-content/uploads/2025/07/impflogo.jpgrgraeber2020-12-23 13:17:332024-06-14 15:51:31Coronaimpfung – Dies sollten Sie beachten!
Erwartet britische Arzneimittelbehörde massive Nebenwirkungen der Covid-19 Impfstoffe?
Ja, sogar massive Nebenwirkungen. Diese Aussage findet man aber nur sehr indirekt und zwar in einem Dokument der britischen Aufsichts- und Zulassungsbehörde für Arzneimittel „Medicines and Healthcare products Regulatory Agency“ (MHRA). Sie ist dort unter anderem zuständig für die Risikoüberwachung jeglicher Medizinprodukte.
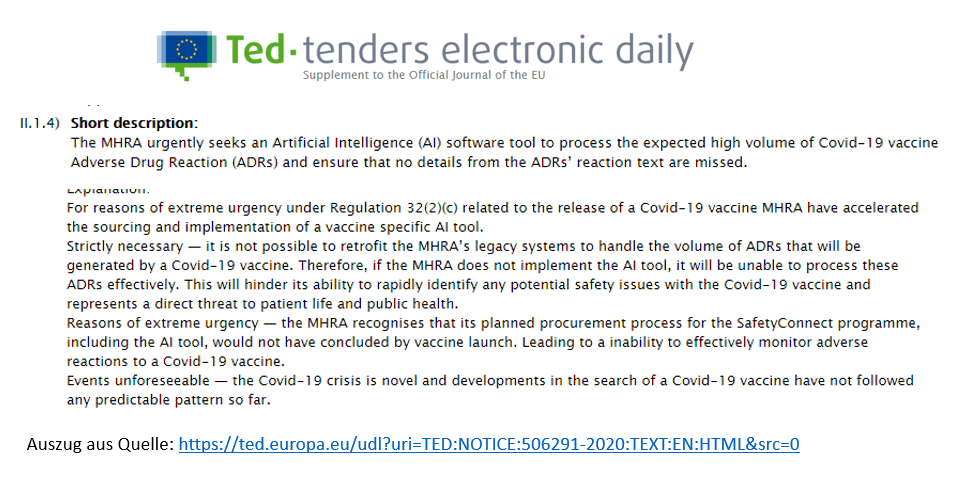
Die obige Grafik entstammt einer Seite der europäischen Union, auf der die EU-weiten öffentlichen Aufträge einzusehen sind. Diese Webseite heißt „Tenders Electronic Daily“ (TED), was man mit „tägliche elektronische Ausschreibungen“ übersetzen kann. Die Einrichtung gehört dem „Amt für Veröffentlichungen der EU“ an. Dort lohnt es sich, mal nach der Ausschreibung Nummer 506291-2020 zu suchen.
Darin geht es um den Auftrag, eine spezielle, neue KI-Software (KI = künstliche Intelligenz) zu erstellen, mit der es möglich sein wird, die unüberschaubare Fülle an gemeldeten Nebenwirkungen durch Impfungen gegen COVID-19 zu erfassen und einer Auswertung zuführen zu können. Die Begründung dafür, warum dies so schnell wie möglich in diesem Umfang und auf diesem Qualitätsniveau erforderlich ist, finden wir schwarz auf weiß bei der MHRA, siehe oben.
Die Erklärung lässt sich in etwa so übersetzen:
Aufgrund der extrem dringlichen Regulierung 32(2)(c) in Bezug auf die Ausgabe eines COVID-19-Impfstoffes will die MHRA in einem beschleunigten Verfahren die Programmierung und Implementierung eines KI-gestützten Tools mit einem Fokus auf Impfstoffe vorantreiben.
Was dazu dringend zu beachten ist:
Es ist nicht möglich, das bisherige System der MHRA nachzurüsten, da die zu erwartende Menge an Berichten über unerwünschte Nebenwirkungen die bisherige Architektur völlig überfordern wird. Daher sieht sich die MHRA gezwungen, ein neuartiges KI-gestütztes System zu implementieren, um wirklich alle Meldungen zu Nebenwirkungen effizient verarbeiten zu können.
Das erklärte Ziel ist hier die Fähigkeit zur schnellen Identifizierung jeglichen potenziellen Sicherheitsrisikos in Verbindung mit COVID-19-Impfstoffen, um eine direkte Gefährdung des Lebens von Patienten beziehungsweise der öffentlichen Gesundheit auszuschließen.
Die Gründe der extremen Dringlichkeit bestehen darin, dass sich die MHRA sehr wohl darüber im Klaren ist, dass der geplante Aufbauprozess für das „SafetyConnect“ Programm einschließlich des KI-Tools bis zur Auslieferung der ersten Impfdosen noch nicht abgeschlossen werden kann, was in der Folge zu einer starken Beeinträchtigung eines belastbaren Monitorings aller Nebenwirkungen von COVID-19-Impfstoffen führt.
Mögliche unvorhersehbare Ereignisse – Die COVID-19-Krise ist ein Novum und die Entwicklungen im Rahmen der Suche nach COVID-19-Impfstoffen konnten keinem vorgegebenen Muster (Blaupause) folgen.
Diese Ausschreibung zeigt indirekt, dass man mit massiven Nebenwirkungen bezüglich der neuartigen Impfstoffe zu rechnen scheint und dafür ein neues Software-Tool benötigt um diese auch alle erfassen zu können.
Übrigens: Wenn Sie solche Informationen interessieren, dann fordern Sie unbedingt meinen kostenlosen Praxis-Newsletter „Unabhängig. Natürlich. Klare Kante.“ dazu an:
Ich hatte bereits in mehreren Beiträgen darauf hingewiesen, dass die „Jubelberichte“ bezüglich der neuartigen Impfstoffe deutlich überzogen erscheinen: